
Ataxin-2 repeat expansions in ALS
Background
ALS is a fatal neurodegenerative disease characterized by the loss of motor neurons in the motor cortex, brainstem and spinal cord. In most patients, the cause of the disease is unknown, although some cases known as familial ALS are associated with mutations in specific genes. Despite the knowledge acquired on causative genes in ALS, the cause of disease heterogeneity remains largely unexplained. Some studies suggest that this variability is caused by disease-modifying factors (such as genetic risk factors). We are particularly interested in Ataxin-2 (ATXN2), a potential genetic risk factor for ALS. The ATXN2 gene contains repeat expansions that vary in length between individuals. The aim of our project is to understand the role of intermediate ATXN2 repeat expansions in the development and disease severity of ALS.
Methods
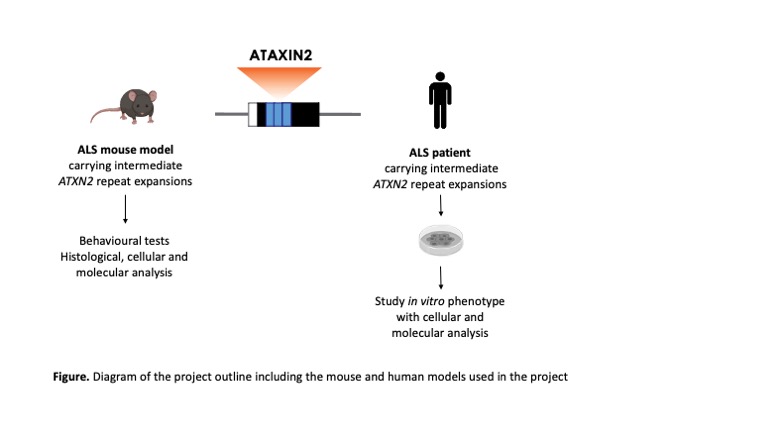
We have generated an ALS mouse model that harbours ALS-associated mutant TDP43 and ATXN2 containing intermediate repeat expansions. We use this model to investigate how ATXN2 contributes to ALS at many levels by performing behavioural tests and histological, cellular and molecular analysis. In addition, we use iPSC-derived from ALS patients that carry intermediate ATXN2 repeat expansions to study the phenotype of iPSC-derived motor neurons and microglia, two of the cell types primarily affected in ALS patients. Ultimately, we want to integrate all these data to better understand how ATXN2 contributes to ALS pathogenesis.
Relevance
Our research will contribute to the understanding of how certain genetic risk factors, in this case ATXN2, modify ALS. This knowledge is essential to find therapeutic targets and develop future treatments that can have disease-modifying effects and therefore increase the lifespan and quality of life of ALS patients.
Team Members
Jeroen Pasterkamp
Marta Cañizares Luna
Financiering: Stichting ALS Nederland; E-rare3 project ‘MAXOMOD’
